Una malattia si definisce "rara" quando la sua prevalenza, intesa come il numero di caso presenti su una data popolazione, non supera una soglia stabilita. In UE la soglia è fissata allo 0,05 per cento della popolazione, non più di 1 caso ogni 2000 persone. Il numero di malattie rare conosciute e diagnosticate è di circa 10.000, ma è una cifra che cresce con l’avanzare della scienza e, in particolare, con i progressi della ricerca genetica. Stiamo dunque parlando non di pochi malati, ma di milioni di persone in Italia e circa 30 milioni in Europa. [Fonte: Eu Commission] Secondo la rete Orphanet Italia, nel nostro Paese i malati rari sono circa 2 milioni: nel 70% dei casi si tratta di pazienti in età pediatrica.
In base ai dati coordinati dal Registro Nazionale Malattie Rare dell'Istituto Superiore di Sanità (ISS), in Italia si stimano 20 casi di malattie rare ogni 10.000 abitanti e ogni anno sono circa 19.000 i nuovi casi segnalati dalle oltre 200 strutture sanitarie diffuse in tutta la penisola. Il 20% delle patologie coinvolge persone in età pediatrica (di età inferiore ai 14 anni). In questa popolazione di pazienti, le malattie rare che si manifestano con maggiore frequenza sono le malformazioni congenite (45%), le malattie delle ghiandole endocrine, della nutrizione o del metabolismo e i disturbi immunitari (20%). Per i pazienti in età adulta, invece, le malattie rare più frequenti appartengono al gruppo delle patologie del sistema nervoso e degli organi di senso (29%) o del sangue e degli organi ematopoietici (18%). [Fonte: ISS 2015]
Vista la mancanza di un’univoca definizione di malattia rara a livello internazionale, ci sono diverse liste di patologie: - National Organization for Rare Disorders (NORD) - Office of Rare Diseases - Orphanet (che propone una lista di circa 6.000 nomi di patologie rare, sinonimi compresi). In Italia, l’Istituto Superiore di Sanità ha individuato un elenco di malattie rare esenti-ticket. Alcune Regioni italiane hanno deliberato esenzioni per patologie ulteriori da quelle previste dal Decreto 279/2001.
L’eventuale approvazione del farmaco in Europa è attesa per la seconda metà del 2021
Milano – L’Agenzia Europea per i Medicinali (EMA) ha accettato di esaminare la richiesta di autorizzazione alla commercializzazione (Marketing Authorization Application, MAA) per avalglucosidasi alfa, come terapia enzimatica sostitutiva di lungo periodo per il trattamento di pazienti con malattia di Pompe. Avalglucosidasi alfa è una terapia enzimatica sostitutiva sperimentale che, se approvata, potrà costituire un potenziale nuovo standard di trattamento per la patologia.
Nicola Sverzellati (Parma): “Nelle persone con IPF o sclerosi sistemica, il virus SARS-CoV-2 avrebbe effetti gravissimi sulla capacità respiratoria, già danneggiata dalla malattia”
Roma – Bersaglio numero uno del COVID-19, anche se non unico, sono i polmoni, quei due organi elastici che contraendosi permettono l’ossigenazione attraverso un continuo scambio di gas tra corpo e ambiente esterno. A minare questa loro vitale capacità possono essere però anche dellemalattie rare, ad esempio quellefibrosanti, che, causando la sostituzione del normale tessuto polmonare con tessuto cicatriziale, li rendono rigidi al punto da non riuscire ad assorbire l'ossigeno presente nell'aria: la fibrosi polmonare idiopatica (IPF) o la sclerosi sistemica, che nel 90% dei casi colpisce anche i polmoni, sono tra queste.
La terapia genica è progettata per interrompere l’accumulo tossico di acidi grassi a catena molto lunga nel cervello
Zurigo (SVIZZERA) – L’Agenzia Europea per i Medicinali (EMA) ha accettato in questi giorni la domanda di autorizzazione all’immissione in commercio (MAA) della terapia genica sperimentale elivaldogene autotemcel (eli-cel) per il trattamento di un sottogruppo di pazienti con adrenoleucodistrofia cerebrale (CALD), una malattia neurodegenerativa letale che colpisce principalmente i giovani maschi.
Premiati Pasquale Agosti e Roberta Gualtierotti (Policlinico Milano) e il prof. Mirko Pinotti (Università di Ferrara)
Milano – Emofilia: grande riconoscimento per la ricerca italiana. Sono ben tre, infatti, i medici italiani premiati al Bayer Hemophilia Awards Program (BHAP), il programma che sostiene progetti di ricerca clinica e di base, oltre ad iniziative educazionali in emofilia in tutto il mondo. I premiati sono Pasquale Agosti e Roberta Gualtierotti, medici specialisti della U.O.C. Medicina Generale - Emostasi e Trombosi presso la Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico di Milano e Mirko Pinotti, Professore Associato del Dipartimento di Scienze della Vita e Biotecnologie dell’Università di Ferrara.
“Homes not hospitals” è il tema di quest’anno: terapia domiciliare e consapevolezza, le sfide del futuro oltre la pandemia
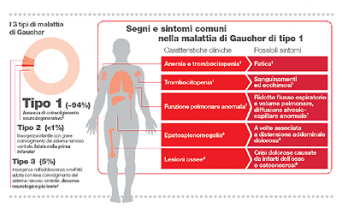
Roma – Il 1° ottobre si celebra la Giornata Mondiale della Malattia di Gaucher, patologia ereditaria autosomica recessiva, per la quale al momento non esistono cure risolutive ma terapie che contribuiscono a curare molti dei sintomi principali, come ingrossamento della milza e del fegato, lesioni ossee, rischio di sanguinamenti e una funzione polmonare anomala. Con un’incidenza di 1:50.000/100.000, la malattia di Gaucher è la più comune tra le 50 patologie da accumulo lisosomiale. È provocata dalla carenza dell’enzima glucocerebrosidasi, normalmente deputato a dissolvere i glicosfingolipidi che, accumulandosi progressivamente nel fegato, nella milza e nel midollo osseo ne alterano la funzione fino a un danno irreversibile.
Il racconto del giovane rumeno: “Dopo 7 anni senza terapia, in Italia ho iniziato a fare la plasmaferesi e, grazie alla lomitapide, ho potuto dimezzarne la frequenza”
Roma – Andrei ha 7 anni, quando in seguito a un controllo di routine scopre di avere il colesterolo totale a livelli spaventosamente alti, 1.100 mg/dL. È l'effetto di una malattia genetica silenziosa, che agisce molto in fretta e, se non trattata tempestivamente, può arrecare danni gravi e irreversibili: l'ipercolesterolemia familiare in forma omozigote. Il bambino non ha alcun sintomo che possa far sospettare la malattia: l'unico segno visibile sono gli xantomi, simili a dei nei di colore giallo/arancione sui gomiti, sulle ginocchia e sui tendini d'Achille, ma compaiono solo dopo gli 8 anni, quando la patologia è già stata individuata casualmente.
L’intervista di OMaR ad Alessandro Guida, regista della pellicola ispirata a una storia vera
Pietro fa lo scout, ma non gli lasciano fare le escursioni; Vittorio frequenta il catechismo, ma all’oratorio non lo fanno giocare a calcetto. Sono loro, e altri come loro, i protagonisti di “I miei supereroi”, il cortometraggio che, in soli sette minuti, punta a raccontare le speranze e le fragilità, l’incertezza e il coraggio di un gruppo di bambini che convivono con l’emofilia, una malattia rara ed ereditaria del sangue, che si manifesta solo nei maschi e che in Italia colpisce circa 4.000 persone.


















Seguici sui Social