Una malattia si definisce "rara" quando la sua prevalenza, intesa come il numero di caso presenti su una data popolazione, non supera una soglia stabilita. In UE la soglia è fissata allo 0,05 per cento della popolazione, non più di 1 caso ogni 2000 persone. Il numero di malattie rare conosciute e diagnosticate è di circa 10.000, ma è una cifra che cresce con l’avanzare della scienza e, in particolare, con i progressi della ricerca genetica. Stiamo dunque parlando non di pochi malati, ma di milioni di persone in Italia e circa 30 milioni in Europa. [Fonte: Eu Commission] Secondo la rete Orphanet Italia, nel nostro Paese i malati rari sono circa 2 milioni: nel 70% dei casi si tratta di pazienti in età pediatrica.
In base ai dati coordinati dal Registro Nazionale Malattie Rare dell'Istituto Superiore di Sanità (ISS), in Italia si stimano 20 casi di malattie rare ogni 10.000 abitanti e ogni anno sono circa 19.000 i nuovi casi segnalati dalle oltre 200 strutture sanitarie diffuse in tutta la penisola. Il 20% delle patologie coinvolge persone in età pediatrica (di età inferiore ai 14 anni). In questa popolazione di pazienti, le malattie rare che si manifestano con maggiore frequenza sono le malformazioni congenite (45%), le malattie delle ghiandole endocrine, della nutrizione o del metabolismo e i disturbi immunitari (20%). Per i pazienti in età adulta, invece, le malattie rare più frequenti appartengono al gruppo delle patologie del sistema nervoso e degli organi di senso (29%) o del sangue e degli organi ematopoietici (18%). [Fonte: ISS 2015]
Vista la mancanza di un’univoca definizione di malattia rara a livello internazionale, ci sono diverse liste di patologie: - National Organization for Rare Disorders (NORD) - Office of Rare Diseases - Orphanet (che propone una lista di circa 6.000 nomi di patologie rare, sinonimi compresi). In Italia, l’Istituto Superiore di Sanità ha individuato un elenco di malattie rare esenti-ticket. Alcune Regioni italiane hanno deliberato esenzioni per patologie ulteriori da quelle previste dal Decreto 279/2001.
L'iperfagia nei bambini e negli adolescenti è un segnale da non sottovalutare. Nelle donne, la terapia migliora ovaio policistico e fertilità
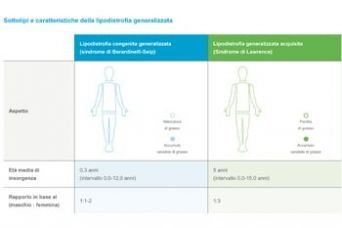
Milano – Si chiamano sindrome di Berardinelli, di Lawrence e di Barraquer-Simmons dal nome dei loro scopritori e sono disturbi caratterizzati dalla perdita di grasso localizzata, di origine genetica o acquisita, raggruppati nella definizione di “sindrome lipodistrofica”. Sono le forme di lipodistrofia sotto la lente di ingrandimento dei lavori del CUEM 2019. Le lipodistrofie sono un insieme eterogeneo di disturbi contraddistinti dalla perdita di tessuto adiposo in assenza di privazioni nutrizionali o stati catabolici. Sono classificate in quattro grandi categorie, e la terminologia corretta e’ di estrema importanza dato che ognuna ha caratteristiche precise internazionalmente riconosciute: la forma congenita generalizzata, quella familiare parziale, la generalizzata acquisita e la parziale acquisita, con alcune sotto categorie che simulano la progeria, la malattia genetica che invecchia precocemente chi ne è colpito.
Le priorità sono la fisioterapia e l'assistenza nell'ambito del pronto soccorso, ma anche le attività sportive
Roma – Nel corso dei decenni, la vita degli emofilici è radicalmente cambiata, e con essa gli obiettivi delle associazioni a loro tutela. Oggi i pazienti possono guardare al futuro con più ottimismo, e grazie ai nuovi farmaci che riducono la frequenza delle infusioni, sono in grado di condurre una vita praticamente normale. Le priorità, per le Onlus, sono ora diventate la sensibilizzazione nei confronti dei medici e dei politici, la raccolta di fondi da devolvere alla ricerca e la messa a punto di progetti in ambito sociale, sportivo e culturale che possano migliorare ancora di più la qualità di vita dei malati e delle loro famiglie.
Dal Trentino alla Sicilia, quattro Onlus fondate negli anni bui dello scandalo degli emoderivati ora guardano al futuro, con tanti progetti e iniziative
Roma – Gran parte delle associazioni che oggi sostengono le persone affette da emofilia sono nate negli anni '70. Non è un caso: quel decennio – e tutto il periodo successivo, fino ad arrivare agli anni '90 – ha rappresentato un vero e proprio incubo per una generazione di emofilici. Lo scandalo del sangue infetto e dei suoi derivati, che fece ammalare di epatite B, C e HIV migliaia di persone, e provocò la morte di tante altre, resta impresso nella nostra memoria collettiva. Solo nel 1992, con la legge n. 210, lo Stato riconobbe un indennizzo economico a tutti coloro che avevano contratto i virus. Per molti di loro era già troppo tardi, ma gli altri unirono le forze per rivendicare i loro diritti nei numerosi processi degli anni a seguire.
L’Associazione Emofilici di Brescia (AEB) “E. Ravasio Passeri” Onlus, regolarmente tesserata nella Federazione delle Associazioni Emofilici (FedEmo), è una delle associazioni dedicate ai pazienti emofilici presenti sul territorio italiano. Fino a qualche decina di anni fa, essere affetti da emofilia voleva dire vivere affrontando grosse difficoltà e problematiche, sia dal punto di vista della salute che sociale e lavorativo. Negli anni le terapie a disposizione dei pazienti, che solo in Italia sono circa 5000, sono migliorate fino ad arrivare alle attuali profilassi e ai nuovi farmaci.
Riuniti a Trieste i massimi esperti della patologia, per discutere di trattamenti, cure personalizzate e accesso ai farmaci
Trieste – E' iniziato ieri, presso il Savoia Excelsior Palace di Trieste, il convegno “Emofilia. La certezza della cura”, un grande evento rivolto alla comunità medico-scientifica e ai pazienti sui temi più attuali nel campo dell’emofilia. In una due-giorni dal programma intenso e variegato, relatori e ospiti italiani e internazionali si confronteranno sulle terapie, sull’approccio integrato e personalizzato, sul ruolo sempre più cruciale del paziente e, infine, sul tema dell’accesso alle cure a livello globale.
In Italia, si calcola che siano circa 60 i nuovi casi della patologia ogni anno, per un totale di 700 pazienti, ma si tratta di un numero ampiamente sottostimato
Roma - Un’intera giornata dedicata a pazienti, familiari, medici e operatori della salute del Lazio, riuniti tutti insieme per confrontarsi e approfondire le tematiche riguardanti la malattia di Fabry: è l’Incontro regionale medici-pazienti del Lazio, organizzato da AIAF Onlus, che si è svolto il 22 giugno scorso a Roma. Una delle tappe del circuito di incontri regionali che l’Associazione Italiana di riferimento per i pazienti Anderson-Fabry sta promuovendo in tutta la Penisola con il supporto del suo Comitato Scientifico, il cui obiettivo è di rappresentare un punto di incontro tra pazienti, medici specialisti dei vari Centri regionali, infermieri, ricercatori e tutti i portatori di interesse che si occupano in diversi ambiti di malattia di Fabry.
I primi risultati sono attesi per la fine del 2019. Già resi noti, invece, i dati finali del trial di Fase I/II, che evidenziano una riduzione dell'ossalato pari al 75% rispetto al basale
Cambridge (U.S.A.) – Lo studio di Fase III ILLUMINATE-A sul farmaco lumasiran è pronto a partire: come ha annunciato l'azienda farmaceutica statunitense Alnylam, il trial verrà condotto in 16 siti presenti in otto Paesi, nei quali l'arruolamento dei pazienti è stato appena completato. Il lumasiran è una terapia RNAi sperimentale mirata all'enzima glicolato ossidasi per il trattamento di adulti e bambini con iperossaluria primitiva di tipo 1 (PH1).















Seguici sui Social